Distance / dissimilarity between samples.
Usage
bdiv_table(
biom,
bdiv = "bray",
weighted = NULL,
normalized = NULL,
tree = NULL,
md = ".all",
within = NULL,
between = NULL,
delta = ".all",
norm = "none",
pseudocount = NULL,
power = 1.5,
alpha = 0.5,
transform = "none",
ties = "random",
seed = 0,
cpus = n_cpus(),
...
)
bdiv_matrix(
biom,
bdiv = "bray",
weighted = NULL,
normalized = NULL,
tree = NULL,
within = NULL,
between = NULL,
norm = "none",
pseudocount = NULL,
power = 1.5,
alpha = 0.5,
transform = "none",
ties = "random",
seed = 0,
cpus = n_cpus()
)
bdiv_distmat(
biom,
bdiv = "bray",
weighted = NULL,
normalized = NULL,
tree = NULL,
within = NULL,
between = NULL,
norm = "none",
pseudocount = NULL,
power = 1.5,
alpha = 0.5,
transform = "none",
ties = "random",
seed = 0,
cpus = n_cpus()
)Arguments
- biom
An rbiom object, or any value accepted by
as_rbiom().- bdiv
Beta diversity distance algorithm(s) to use. Options are:
c("aitchison", "bhattacharyya", "bray", "canberra", "chebyshev", "chord", "clark", "sorensen", "divergence", "euclidean", "generalized_unifrac", "gower", "hamming", "hellinger", "horn", "jaccard", "jensen", "jsd", "lorentzian", "manhattan", "matusita", "minkowski", "morisita", "motyka", "normalized_unifrac", "ochiai", "psym_chisq", "soergel", "squared_chisq", "squared_chord", "squared_euclidean", "topsoe", "unweighted_unifrac", "variance_adjusted_unifrac", "wave_hedges", "weighted_unifrac"). For the UniFrac family, a phylogenetic tree must be present inbiomor explicitly provided viatree=. Supports partial matching. Multiple values are allowed for functions which return a table or plot. Default:"bray"- weighted
(Deprecated - weighting is now inherent in bdiv metric name.) Take relative abundances into account. When
weighted=FALSE, only presence/absence is considered. Multiple values allowed. Default:NULL- normalized
(Deprecated - normalization is now inherent in bdiv metric name.) Only changes the "Weighted UniFrac" calculation. Divides result by the total branch weights. Default:
NULL- tree
A
phyloobject representing the phylogenetic relationships of the taxa inbiom. Only required when computing UniFrac distances. Default:biom$tree- md
Dataset field(s) to include in the output data frame, or
'.all'to include all metadata fields. Default:'.all'- within, between
Dataset field(s) for intra- or inter- sample comparisons. Alternatively, dataset field names given elsewhere can be prefixed with
'=='or'!='to assign them towithinorbetween, respectively. Default:NULL- delta
For numeric metadata, report the absolute difference in values for the two samples, for instance
2instead of"10 vs 12". Default:TRUE- norm
Normalize the incoming counts. Options are:
'none': No transformation.'percent': Relative abundance (sample abundances sum to 1).'binary': Unweighted presence/absence (each count is either 0 or 1).'clr': Centered log ratio.
Default:
'none'.- pseudocount
Value added to counts to handle zeros when
norm = 'clr'. Ignored for other normalization methods. Default:NULL(emits a warning).- power
Scaling factor for the magnitude of differences between communities (\(p\)) when
bdiv = 'minkowski'. Ignored for other beta diversity metrics. Default:1.5- alpha
The alpha term to use in Generalized UniFrac. How much weight to give to relative abundances; a value between 0 and 1, inclusive. Setting
alpha=1is equivalent to Normalized UniFrac. Default:0.5- transform
Transformation to apply to calculated values. Options are:
c("none", "rank", "log", "log1p", "sqrt", "percent")."rank"is useful for correcting for non-normally distributions before applying regression statistics. Default:"none"- ties
When
transform="rank", how to rank identical values. Options are:c("average", "first", "last", "random", "max", "min"). Seerank()for details. Default:"random"- seed
Random seed for permutations. Must be a non-negative integer. Default:
0- cpus
The number of CPUs to use. Set to
NULLto use all available, or to1to disable parallel processing. Default:NULL- ...
Not used.
Value
bdiv_matrix()-An R matrix of samples x samples.
bdiv_distmat()-A dist-class distance matrix.
bdiv_table()-A tibble data.frame with columns named .sample1, .sample2, .bdiv, .distance, and any fields requested by
md. Numeric metadata fields will be returned asabs(x - y); categorical metadata fields as"x","y", or"x vs y".
Metadata Comparisons
Prefix metadata fields with == or != to limit comparisons to within or
between groups, respectively. For example, stat.by = '==Sex' will
run calculations only for intra-group comparisons, returning "Male" and
"Female", but NOT "Female vs Male". Similarly, setting
stat.by = '!=Body Site' will only show the inter-group comparisons, such
as "Saliva vs Stool", "Anterior nares vs Buccal mucosa", and so on.
The same effect can be achieved by using the within and between
parameters. stat.by = '==Sex' is equivalent to
stat.by = 'Sex', within = 'Sex'.
See also
Other beta_diversity:
bdiv_boxplot(),
bdiv_clusters(),
bdiv_corrplot(),
bdiv_heatmap(),
bdiv_ord_plot(),
bdiv_ord_table(),
bdiv_stats(),
distmat_stats()
Examples
library(rbiom)
# Subset to four samples
biom <- hmp50$clone()
biom$counts <- biom$counts[,c("HMP18", "HMP19", "HMP20", "HMP21")]
# Return in long format with metadata
bdiv_table(biom, 'w_unifrac', md = ".all")
#> # A tibble: 6 × 8
#> .sample1 .sample2 .bdiv .distance Age BMI `Body Site` Sex
#> <chr> <chr> <fct> <dbl> <dbl> <dbl> <fct> <fct>
#> 1 HMP18 HMP19 w_unifrac 0.665 0 3 Saliva vs Stool Female vs M…
#> 2 HMP18 HMP20 w_unifrac 0.681 1 2 Saliva vs Stool Female vs M…
#> 3 HMP18 HMP21 w_unifrac 0.717 4 2 Saliva vs Stool Male
#> 4 HMP19 HMP20 w_unifrac 0.418 1 5 Stool Female
#> 5 HMP19 HMP21 w_unifrac 0.390 4 1 Stool Female vs M…
#> 6 HMP20 HMP21 w_unifrac 0.149 5 4 Stool Female vs M…
# Only look at distances among the stool samples
bdiv_table(biom, 'w_unifrac', md = c("==Body Site", "Sex"))
#> # A tibble: 3 × 6
#> .sample1 .sample2 .bdiv .distance `Body Site` Sex
#> <chr> <chr> <fct> <dbl> <fct> <fct>
#> 1 HMP19 HMP20 w_unifrac 0.418 Stool Female
#> 2 HMP19 HMP21 w_unifrac 0.390 Stool Female vs Male
#> 3 HMP20 HMP21 w_unifrac 0.149 Stool Female vs Male
# Or between males and females
bdiv_table(biom, 'w_unifrac', md = c("Body Site", "!=Sex"))
#> # A tibble: 4 × 6
#> .sample1 .sample2 .bdiv .distance `Body Site` Sex
#> <chr> <chr> <fct> <dbl> <fct> <fct>
#> 1 HMP18 HMP19 w_unifrac 0.665 Saliva vs Stool Female vs Male
#> 2 HMP18 HMP20 w_unifrac 0.681 Saliva vs Stool Female vs Male
#> 3 HMP19 HMP21 w_unifrac 0.390 Stool Female vs Male
#> 4 HMP20 HMP21 w_unifrac 0.149 Stool Female vs Male
# All-vs-all matrix
bdiv_matrix(biom, 'w_unifrac')
#> HMP18 HMP19 HMP20 HMP21
#> HMP18 0.0000000 0.6651627 0.6810017 0.7170374
#> HMP19 0.6651627 0.0000000 0.4183059 0.3896741
#> HMP20 0.6810017 0.4183059 0.0000000 0.1490926
#> HMP21 0.7170374 0.3896741 0.1490926 0.0000000
#> attr(,"cmd")
#> [1] "bdiv_matrix(biom, \"w_unifrac\", cpus = 4)"
# All-vs-all distance matrix
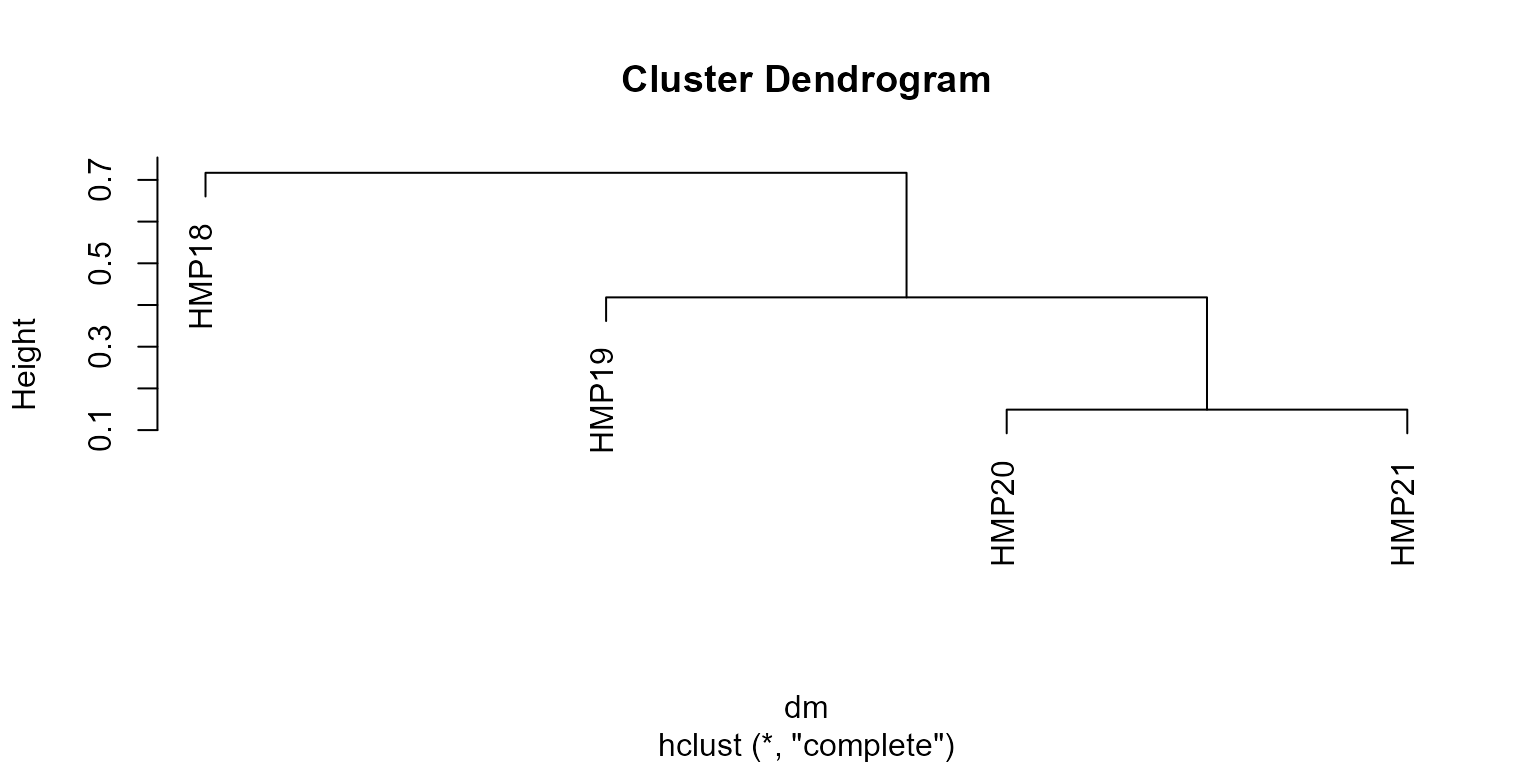
dm <- bdiv_distmat(biom, 'w_unifrac')
dm
#> HMP18 HMP19 HMP20
#> HMP19 0.6651627
#> HMP20 0.6810017 0.4183059
#> HMP21 0.7170374 0.3896741 0.1490926
plot(hclust(dm))