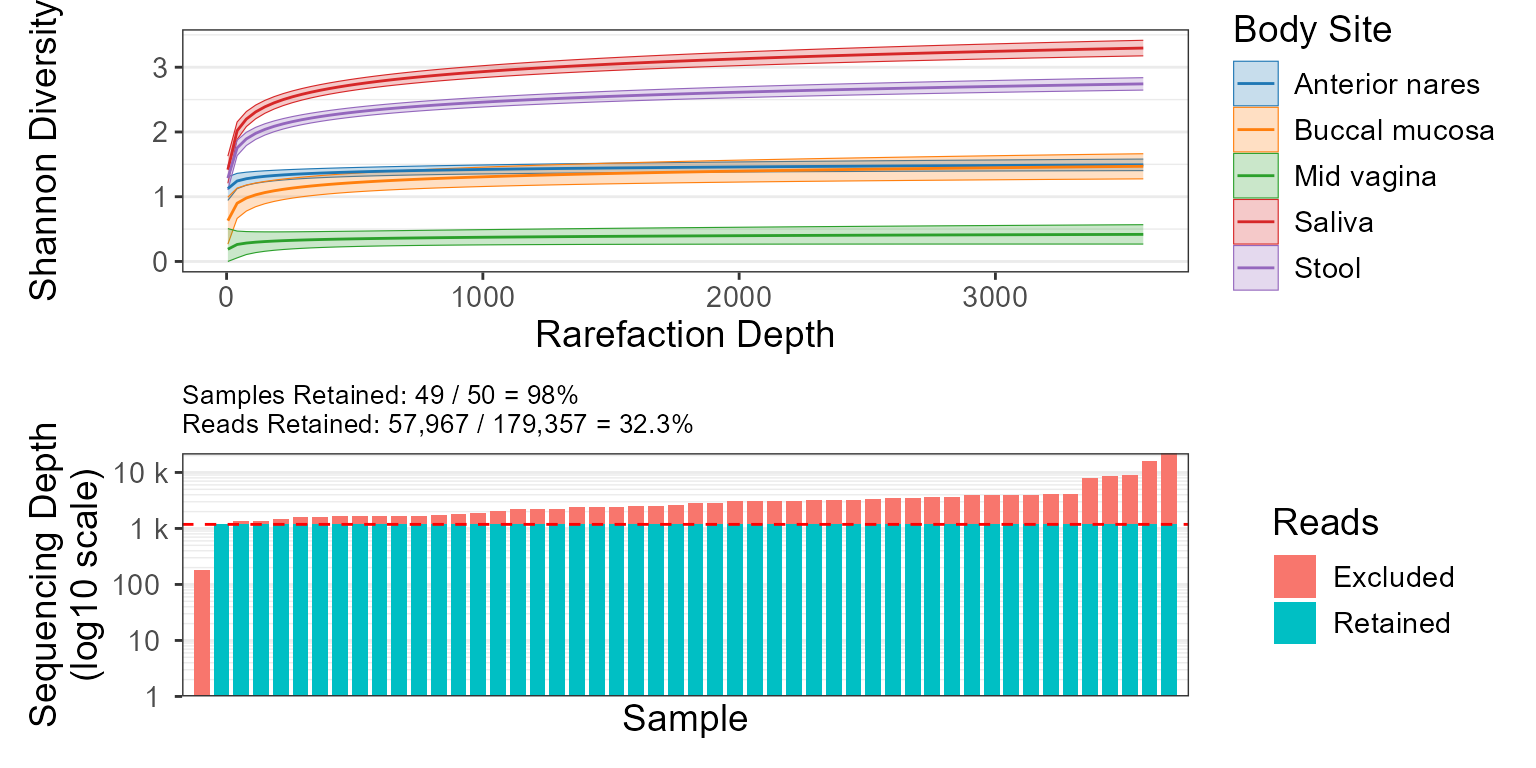
Combines rare_corrplot and rare_stacked into a single figure.
Source:R/rare_multiplot.r
rare_multiplot.RdCombines rare_corrplot and rare_stacked into a single figure.
Usage
rare_multiplot(
biom,
adiv = "Shannon",
layers = "tc",
rline = TRUE,
stat.by = NULL,
facet.by = NULL,
colors = TRUE,
shapes = TRUE,
test = "none",
fit = "log",
at = NULL,
level = 0.95,
p.adj = "fdr",
transform = "none",
alt = "!=",
mu = 0,
caption = TRUE,
check = FALSE,
...
)Arguments
- biom
An rbiom object, or any value accepted by
as_rbiom().- adiv
Alpha diversity metric(s) to use. Options are:
c("ace", "berger", "brillouin", "chao1", "faith", "fisher", "simpson", "inv_simpson", "margalef", "mcintosh", "menhinick", "observed", "shannon", "squares"). For"faith", a phylogenetic tree must be present inbiomor explicitly provided viatree=. Setadiv=".all"to use all metrics. Multiple/abbreviated values allowed. Default:"shannon"- layers
One or more of
c("trend", "confidence", "point", "name", "residual"). Single letter abbreviations are also accepted. For instance,c("trend", "point")is equivalent toc("t", "p")and"tp". Default:"tc"- rline
Where to draw a horizontal line on the plot, intended to show a particular rarefaction depth. Set to
TRUEto show an auto-selected rarefaction depth orFALSEto not show a line. Default:NULL- stat.by
Dataset field with the statistical groups. Must be categorical. Default:
NULL- facet.by
Dataset field(s) to use for faceting. Must be categorical. Default:
NULL- colors
How to color the groups. Options are:
TRUE-Automatically select colorblind-friendly colors.
FALSEorNULL-Don't use colors.
- a palette name -
Auto-select colors from this set. E.g.
"okabe"- character vector -
Custom colors to use. E.g.
c("red", "#00FF00")- named character vector -
Explicit mapping. E.g.
c(Male = "blue", Female = "red")
See "Aesthetics" section below for additional information. Default:
TRUE- shapes
Shapes for each group. Options are similar to
colors's:TRUE,FALSE,NULL, shape names (typically integers 0 - 17), or a named vector mapping groups to specific shape names. See "Aesthetics" section below for additional information. Default:TRUE- test
Method for computing p-values:
'none','emmeans', or'emtrends'. Default:'emmeans'- fit
How to fit the trendline. Options are
'lm','log', and'gam'. Default:'log'- at
Position(s) along the x-axis where the means or slopes should be evaluated. Default:
NULL, which samples 100 evenly spaced positions and selects the position where the p-value is most significant.- level
The confidence level for calculating a confidence interval. Default:
0.95- p.adj
Method to use for multiple comparisons adjustment of p-values. Run
p.adjust.methodsfor a list of available options. Default:"fdr"- transform
Transformation to apply to calculated values. Options are:
c("none", "rank", "log", "log1p", "sqrt", "percent")."rank"is useful for correcting for non-normally distributions before applying regression statistics. Default:"none"- alt
Alternative hypothesis direction. Options are
'!='(two-sided; not equal tomu),'<'(less thanmu), or'>'(greater thanmu). Default:'!='- mu
Reference value to test against. Default:
0- caption
Add methodology caption beneath the plot. Default:
TRUE- check
Generate additional plots to aid in assessing data normality. Default:
FALSE- ...
Additional parameters to pass along to ggplot2 functions. Prefix a parameter name with a layer name to pass it to only that layer. For instance,
p.size = 2ensures only the points have their size set to2.
Value
A ggplot2 plot. The computed data points, ggplot2 command,
stats table, and stats table commands are available as $data,
$code, $stats, and $stats$code, respectively.
Aesthetics
All built-in color palettes are colorblind-friendly. The available
categorical palette names are: "okabe", "carto", "r4",
"polychrome", "tol", "bright", "light",
"muted", "vibrant", "tableau", "classic",
"alphabet", "tableau20", "kelly", and "fishy".
Shapes can be given as per base R - numbers 0 through 17 for various shapes, or the decimal value of an ascii character, e.g. a-z = 65:90; A-Z = 97:122 to use letters instead of shapes on the plot. Character strings may used as well.
See also
Other rarefaction:
rare_corrplot(),
rare_stacked(),
sample_sums()
Other visualization:
adiv_boxplot(),
adiv_corrplot(),
bdiv_boxplot(),
bdiv_corrplot(),
bdiv_heatmap(),
bdiv_ord_plot(),
plot_heatmap(),
rare_corrplot(),
rare_stacked(),
stats_boxplot(),
stats_corrplot(),
taxa_boxplot(),
taxa_corrplot(),
taxa_heatmap(),
taxa_stacked()